Charcot Marie Tooth and Dejerine-Sottas syndrome are groups of diseases that involve the breakdown of the myelin sheath covering nerve axons.
As this myelin sheath breaks down, people who have these disorders suffer nerve damage in the arms and legs—those with Dejerine-Sottas disease may never walk or may lose the ability to walk by the time they are teenagers.
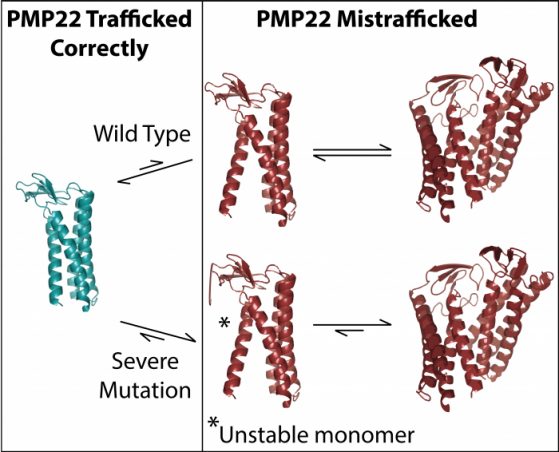
Researchers have known that a protein called PMP22, which is important for nerve myelin, is likely involved in the disease. But because the protein is so small and part of the cell membrane, it’s difficult to study. Now, an interdisciplinary team from the University of Michigan and Vanderbilt University have used a cutting-edge technique called ion mobility-mass spectrometry (IM-MS) to show that an unstable, mutant version of the PMP22 protein associates with another mutant PMP22, forming a stable complex called a “dimer.”
“If you get one of these mutations in your PMP22 gene, that leads to the creation of this mutant protein. Then, our results indicate that the mutant protein produced is likely going to dimerize,” said Brandon Ruotolo, lead author on the new study and a professor of chemistry at U-M. “Because it’s stable, it’s not going to re-form into monomeric protein that is then going to give you normal myelin sheath production.”
Their results, published in the Proceedings of the National Academy of Sciences, provide a new view into how these diseases work, and could one day provide a target for therapeutics.
Vanderbilt researcher Charles Sanders began studying PMP22 in 2000. It was a protein known to be involved in these neurological diseases, but few were studying it. There are other diseases related to problems with protein folding, Sanders says. Typically, misfolding proteins cause disease in two ways: one is that when they misfold, they are degraded and can’t complete the job they’re meant to do. A second is that a protein clumps up, gumming up the cell and preventing it from doing its job properly.
“With PMP22, it’s never been completely clear how it causes disease. It’s definitely related to problems with folding, but how does this really work? What Brandon’s results suggest is that when this protein is unstable, it doesn’t fold correctly, but instead of getting degraded, it forms this misfolded dimer,” Sanders said. “We still don’t know exactly what it then does to cause these diseases, but it’s an interesting and unusual observation that makes these diseases different from all of the other protein-folding diseases.”
Over the years, Sanders’ lab has done a series of measurements to quantitate the stability of mutant forms of PMP22. His lab has also collaborated with U-M researcher Melanie Ohi to study the healthy function of PMP22, work that started while Ohi was on the faculty at Vanderbilt.
“We were able to see what we called myelin-like assemblies that allowed us to get some idea of what this protein might actually be doing in the context of a membrane. When we came to U-M, our goal was to get a high-resolution structure, which is really hard because this is a really small protein,” Ohi said. “We needed to figure out if the protein Dr. Yadav was making for structural studies was forming larger oligomers and, surprisingly, PMP22 works well for Brandon’s technique.”
Currently an associate professor in U-M’s Life Sciences Institute, Ohi met Ruotolo, whose lab specializes in using IM-MS to capture native structures of biomolecules as well as how they assemble.
To view these proteins using this kind of mass spectrometry, Ruotolo’s lab transfers the protein into the gas phase using a technique called nano-electrospray ionization. The technique works by aersolzing protein-containing solutions into tiny charged droplets. The solvent carrying the protein quickly dries—this process happens over just milliseconds—which leaves the charged protein behind. These charged proteins are then drawn into a vacuum environment inside the mass spectrometry instrument.
Once inside the instrument, ions encounter a chamber pressurized with a small amount of background gas. Within this chamber, the instrument uses an electric field to pull the ionized proteins through a series of electrodes in the presence of the background gas. This separates the ions according to their size. A subsequent device uses a chamber operated under strict vacuum conditions to separate the same ions according to their mass. This way, the researchers are able to view both the structures of the proteins, and separate PMP22 dimers from monomers in order to evaluate them independently.
The researchers then heat the ionized proteins and measure their size, repeating this step several times in order to measure the changes in size as the proteins unfold. This tells the researchers how stable the proteins are—and in particular, the stability of the misfolded dimer proteins.
The researchers say developing therapeutics to interfere with these mutants clumping together is further down the road, but knowing how and where the protein self-associates provides a road map for a new venue of research.
“Not only is this a big step forward in what we know about the protein, mass spectrometry has such fabulous capabilities that this opens the door for a whole unbelievable set of things that we could do in the future with this protein,” Sanders said. “With the sort of techniques that Brandon works with, you could imagine eventually extending this to drug discovery, using this basic method as an assay for finding compounds that correct these defects.”
The study is titled “Ion Mobility Mass Spectrometry Reveals the Role of Peripheral Myelin Protein Dimers in Peripheral Neuropathy.” PNAS has made it available at DOI:10.1073/pnas.2015331118.
Study co-authors include U-M chemistry graduate students Sarah Fantin, Kristine Parson and Brock Juliano; Life Sciences Institute senior research associate Pramod Yadav; and Vanderbilt research fellow Geoffrey Li.
-- Morgan Sherburne, UM News